Options
Query and background samples
Composition Profiler provides an easy way to visually investigate bias in amino acid composition between two sets of protein sequences. A set of proteins under study (the query sample) can be analyzed against a representative sample of proteins from the organism under study, or a group of proteins with a contrasting functional annotation (the background sample), which provides a suitable background amino acid distribution.
Sequences should be entered in FastA format.
There are no theoretical limits on the number of sequences that can be given as input. Composition Profiler treats differences in amino acids composition as "signals" – since the p-value is a function of the sample size and signal strength, samples which are not large enough for the inherent signal will give differences with p-value above the statistical significance threshold, and will be discarded as spurious. With small sample sizes, only the strongest signals will be identified.
For example, if a sample consisting of only one sequence, AAAAAAAAAA, were to be analyzed against SwissProt, because the difference between 100% A in the sample and 7.89% A in SwissProt presents a very strong signal (12 fold enrichment), the difference will be statistically significant. Larger sample size allows identification of more subtle signals.
Background distribution
Alternatively, the query sample can be analyzed against one of the standard protein datasets:
- SwissProt 51 (Bairoch et al., 2005), closest to the distribution of amino acids in nature among the four datasets
- PDB Select 25 (Berman et al, 2000), a subset of structures from the Protein Data Bank with less than 25% sequence identity, biased towards the composition of proteins amenable to crystallization studies
- Surface residues determined by the Molecular Surface Package over a sample of PDB structures of monomeric proteins, suitable for analyzing phenomena on protein surfaces, such as binding
- DisProt 3.4, comprised of a set of consensus sequences of experimentally determined disordered regions (Sickmeier et al., 2007)
To illustrate the importance of choosing an appropriate background distribution, we generated composition profiles of (A) PDBS25, (B) surface residues dataset and (C) DisProt against SwissProt:
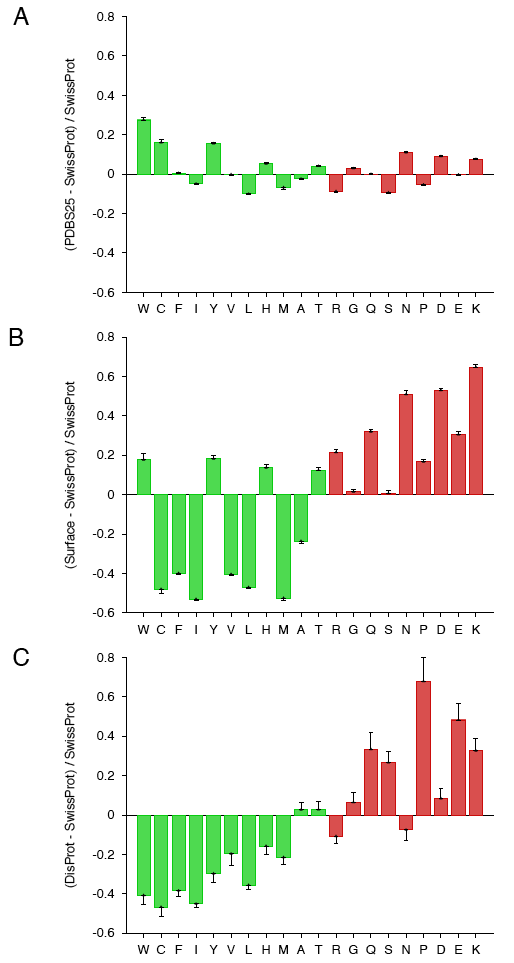
All three graphs have the same y-axis scale, the same order of amino acids and the same color-coding scheme (Vihinen flexibility), which allows direct visual comparison of the amino acid biases in the three datasets.
Discovery
Alpha value
Statistical significance of an observed enrichment or depletion is estimated using permutations. Concatenation of query and backround sequences is randomly permuted and a test statistic more extreme than the one observed in the actual data is counted towards the pvalue.
Any particular enrichment or depletion is statistically significant when the P-value is lower than or equal to a user-specified statistical significance (alpha) value.
Bonferroni correction
Simple multiple testing correction. Assumes independence of tests, and divides the alpha value by the total number of tests. See (Weisstein) for details.
Number of iterations
In the context of calculating composition differences, bootstrapping is used for non-parametric estimation of the confidence intervals for reported amino acid compositions. More precisely, reported confidence intervals are standard deviations of pseudo-replicate compositions.
In the context of discovery and relative entropy calculations, permutations are used to estimate the statistical significance of the observed test statistic (fractional difference or relative entropy). In each iteration, a random permutation of the two starting samples is created, the test statistic is computed based on the permuted sample and permutation test statistic more extreme than the observed test statistic is coutned towards the P-value.
Amino acid grouping, ordering and color-coding
Alpha helix frequency (Nagano, 1973)
Y P G N S R T C I V D W Q L K M F A H E 0.63 0.70 0.72 0.77 0.78 0.83 0.87 0.94 0.94 0.97 1.00 1.06 1.10 1.23 1.23 1.23 1.23 1.29 1.29 1.54
Aromatics
Aromatic amino acids (F, W, Y) are colored green, and the remaining amino acids are colored black.
Beta structure frequency (Nagano, 1973)
E R N P S K H D G A Y C W Q T L M F V I 0.33 0.67 0.72 0.75 0.77 0.81 0.87 0.9 0.9 0.96 1.07 1.13 1.13 1.18 1.23 1.26 1.29 1.37 1.41 1.54
Bulkiness (Zimmerman et al., 1968)
G S A D N C E H R Q K T M P Y F I L V W 3.4 9.47 11.5 11.68 12.82 13.46 13.57 13.69 14.28 14.45 15.71 15.77 16.25 17.43 18.03 19.8 21.4 21.4 21.57 21.67
Charge
Positively charged residues (K, R) are colored blue, negatively charged residues (D, E) are colored red; and neutral residues are colored black.
Coil propensity (Nagano, 1973)
F M L A E H I Q V K W C T D R S G Y N P 0.58 0.62 0.63 0.72 0.75 0.76 0.8 0.81 0.83 0.84 0.87 1.01 1.03 1.04 1.33 1.34 1.35 1.35 1.38 1.43
Disorder propensity (Dunker et al., 2001)
Disorder-promoting residues (A, R, S, Q, E, G, K, P ) are colored red, order-promoting residues (N, C, I, L, F, W, Y, V) are colored blue, and disorder-order neutral residues (D, H, M, T) are colored black.
Flexibility (Vihinen et al., 1994)
W C F I Y V L H M A T R G Q S N P D E K 0.904 0.906 0.915 0.927 0.929 0.931 0.935 0.950 0.952 0.984 0.997 1.008 1.031 1.037 1.046 1.048 1.049 1.068 1.094 1.102
Hydrophobicity (Eisenberg et al., 1984)
R K D Q N E H S T P Y C G A M W L V F I -2.53 -1.5 -0.9 -0.85 -0.78 -0.74 -0.4 -0.18 -0.05 0.12 0.26 0.29 0.48 0.62 0.64 0.81 1.06 1.08 1.19 1.38
Hydrophobicity (Kyte and Doolittle, 1982)
R K D E N Q H P Y W S T G A M C F L V I -4.5 -3.9 -3.5 -3.5 -3.5 -3.5 -3.2 -1.6 -1.3 -0.9 -0.8 -0.7 -0.4 1.8 1.9 2.5 2.8 3.8 4.2 4.5
Hydrophobicity (Fauchere and Pliska, 1983)
R K D E N Q S G H T A P Y V C L F I M W -1.01 -0.99 -0.77 -0.64 -0.6 -0.22 -0.04 0.0 0.13 0.26 0.31 0.72 0.96 1.22 1.54 1.7 1.79 1.8 1.23 2.25
Interface propensity (Jones and Thornton, 1997)
D K S P T A E Q G N R V L H C I M Y F W -0.38 -0.36 -0.33 -0.25 -0.18 -0.17 -0.13 -0.11 -0.07 0.12 0.27 0.27 0.4 0.41 0.43 0.44 0.66 0.66 0.82 0.83
Linker propensity (George and Heringa, 2003)
C G W D I N K S V A Y H T M Q E L F R P 0.778 0.835 0.895 0.916 0.922 0.944 0.944 0.947 0.955 0.964 1.0 1.014 1.017 1.032 1.047 1.051 1.085 1.119 1.143 1.299
Polarity (Zimmerman et al., 1968)
A G I L V F M C P Y T S W N Q K D E H R 0.0 0.0 0.13 0.13 0.13 0.35 1.43 1.48 1.58 1.61 1.66 1.67 2.1 3.38 3.53 49.5 49.7 49.9 51.6 52.0
Size (Dawson, 1972)
G A D C S N E T V I L P Q H M F K W Y R 0.5 2.5 2.5 3.0 3.0 5.0 5.0 5.0 5.0 5.5 5.5 5.5 6.0 6.0 6.0 6.5 7.0 7.0 7.0 7.5
Surface exposure (Janin, 1979)
K R E Q D N Y P T H S A G W M L F V I C -1.8 -1.4 -0.7 -0.7 -0.6 -0.5 -0.4 -0.3 -0.2 -0.1 -0.1 0.3 0.3 0.3 0.4 0.5 0.5 0.6 0.7 0.9
Solvation potential (Jones and Thornton, 1996)
W F I L M Y V C H A G S T P N R Q D E K -0.68 -0.55 -0.49 -0.49 -0.40 -0.32 -0.31 -0.30 -0.06 0.05 0.08 0.15 0.16 0.19 0.22 0.41 0.45 0.64 0.77 1.61
Additional color schemes
WebLogo default colors
Amino Acid Color Name RGB Hexadecimal G, S, T, Y, C green [0,204,0] 00CC00 N, Q purple [204,0,204] CC00CC K, R, H blue [0,204,0] 0000CC D, E red [204,0,0] CC0000 P,A,W,F,L,I,M,V black [0,0,0] 000000
Amino Colors
Amino Acid Color Name RGB Hexadecimal D, E bright red [230,10,10] E60A0A C, M yellow [230,230,0] E6E600 K, R blue [20,90,255] 145AFF S, T orange [250,150,0] FA9600 F, Y mid blue [50,50,170] 3232AA N, Q cyan [0,220,220] 00DCDC G light grey [235,235,235] EBEBEB L, V, I green [15,130,15] 0F820F A dark grey [200,200,200] C8C8C8 W pink [180,90,180] B45AB4 H pale blue [130,130,210] 8282D2 P flesh [220,150,130] DC9682
Shapley color table
Amino Acids Color Name RGB Hexadecimal D, T dark red [160,0,66] A00042 E red-brown [102,0,0] 660000 C bright yellow [255,255,112] FFFF70 M, Y dark yellow [184,160,66] B8A042 K blue [71,71,184] 4747B8 R dark blue [0,0,124] 00007C S, Q orange [255,76,76] FF4C4C F, P, W dark grey [83,76,66] 534C42 N flesh [255,124,112] FF7C70 G, V light grey [235,235,235] EBEBEB I dark green [0,76,0] 004C00 L grey-green [69,94,69] 455E45 A light green [140,255,140] 8CFF8C H pale blue [112,112,255] 7070FF
In the original Shapley scheme, G and V are color-coded as white. Since this would render them invisible against a white background, their color has been changed to light grey.
Show Y-axis label
Shows label on the Y-axis of the profile graph.
Output format
Composition Profiler supports:
- Portable Network Graphics (PNG)
- Portable Document Format (PDF)
- Encapsulated PostScript (EPS)
Additionally, raw values for bar heights and error bars are displayed if plain text (TXT) option is chosen.
Output size
Height and width of the output image, in pixels, centimeters or inches.
Resolution
Sets up the image resolution. Applicable to bitmaps only (PNG).
References
- Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, Ferro S,
Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA,
O'Donovan C, Redaschi N, and Yeh LS. (2005) "The Universal Protein
Resource (UniProt)." Nucleic Acids Research 33:D154-159.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H,
Shindyalov IN, and Bourne PE. (2000) "The Protein Data Bank."
Nucleic Acids Research, 28:235-242.
- Dawson DM. (1972) In The Biochemical Genetics of Man (Brock DJH and Mayo O, eds.), Academic Press, New York, 1-38.
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS,
Oldfield CJ, Campen AM, Ratliff CM, Hipps KW et al.
(2001) "Intrinsically disordered protein." J. Mol. Graph.
Model., 19, 26-59.
- Eisenberg D, Schwarz E, Komaromy M, and Wall R. (1984) "Analysis of
membrane and surface protein sequences with the hydrophobic moment
plot." J. Mol. Biol. 179:125-142.
- Fauchere J-L, and Pliska VE. (1983) "Hydrophobic parameters pi of
amino acid side chains from partitioning of N-acetyl-amino-acid
amides." Eur. J. Med. Chem. 18:369-375.
- George RA, and Heringa J. (2003) An analysis of protein domain
linkers: their classification and role in protein folding. Protein
Eng. 15:871-879.
- Hogg RV and Craig A. (1994) Introduction to Mathematical Statistics, 5th edition, Prentice Hall.
- Janin J. (1979) "Surface and inside volumes in globular proteins."
Nature, 277, 491-492.
- Jones S, and Thornton J. (1997) "Analysis of protein-proteins
interaction sites using surface patches." J. Mol. Biol.
272:121-132.
- Jones S, and Thornton J. (1996) "Principles of protein-protein
interactions." Proc. Natl. Acad. Sci. USA, 93:13-20.
- Kyte J, and Doolittle RF. (1982) "A simple method for displaying the
hydropathic character of a protein." J. Mol. Biol. 157:105-132.
- Nagano K. (1973) "Local analysis of the mechanism of protein folding. I. Prediction of helices, loops, and beta-structures from primary structure." J. Mol. Biol. 75:401-420.
- Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A,
Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, and Dunker AK.
(2007) "DisProt: the Database of Disordered Proteins." Nucleic Acids
Research. 35:D786-93.
- Vihinen M, Torkkila E, and Riikonen P. (1994) "Accuracy of protein
flexibility predictions." Proteins, 19, 141-149.
- Weisstein EW. "Bonferroni Correction." From MathWorld - A Wolfram Web Resource. http://mathworld.wolfram.com/BonferroniCorrection.html
- Zimmerman JM, Eliezer N, and Simha R. (1968) "The characterization
of amino acid sequences in proteins by statistical methods." J J.
Theor. Biol. 21:170-201.